随着3D打印在医疗领域的应用逐渐深化,这项技术也得到了FDA的充分重视。2016年5月,美国FDA首次发布3D打印医疗器材指引草案,为应对不断创新且日新月异的3D打印技术发展,美国FDA于2017年12月5日公告了增材制造医疗器材的技术考虑规范。该规范包含设备设计、功能、产品耐久性测试及品质要求等三维打印医疗产品制造技术指导。

为确保法规的监管方式适合此种独特且创新技术,该规范的建构基于美国食品和药物管理局对100多项使用3D打印的医疗产品进行审查的经验基础,包含有膝盖置换物、植入物、重建颅骨及第一种通过3D打印生产的癫痫药物,该药物比起传统药物具有多孔基质,可加速药物溶解。
FDA注意到,3D打印以其独特的优势适合不同特点的医疗器材的制造:
通用医疗器材设计
增材制造可以实现其他制造方式所无法实现的制造灵活性,然而制造过程中的可靠性还需要严格的追踪,包括测量方式和数据备份。表面处理等其他后处理方式也需要考虑进来,以形成全套的可追踪的质量管理方法。
个性化医疗产品
根据患者自身情况而量身定制的医疗器械可以通过很多中方式制造出来,传统方式也可以实现,3D打印方式也可以实现。然而,3D打印方式具备了明显的制造速度和成本优势。
FDA的指导规范分为两大部分:设计与制造的考虑、医疗器械测试的考虑。关于设计与制造的考虑,主要分为技术考虑,尤其是针对质量体系要求方面的需求点,并且包含了制造过程中的考虑。医疗器械测试的考虑方面重点描述了在进入到510(k),PMA,IDE这些过程中需要考虑的因素。
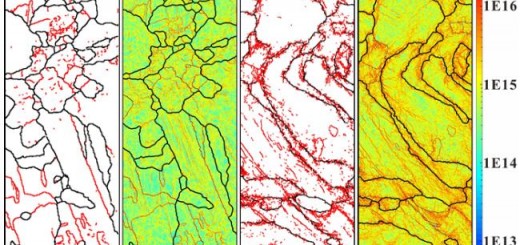
![]() 图像的处理是至关重要的:
图像的处理是至关重要的:
- 图像的影响
用于匹配的最小图像特征质量和分辨率
the minumum image feature quality and resolution used for matching
任何平滑或图像处理算法,当与参考解剖结构相比时,这些算法可以改变最终装置的尺寸。
any smoothing or image processing algorithms that may alter the dimensions of the final device when compared to the reference anatomy.
正在成像的解剖结构的刚性,以及
the rigidity of the anatomic structures being imaged, and
用于将装置与患者解剖结构相匹配的解剖学标志的清晰度。
the clarity of anatomic landmarks used to match the device to the patient’s anatomy.
如果扫描的影像结构不够精确,那么由此生成的三维建模结构就有可能不适合患者,而这些小的变化通常很难觉察到,只有当患者的使用过程中才会发现错位的情况。所以这个过程中,过程验证-Process Validation是十分必要和重要的。FDA也在其增材制造医疗器械规范中有重点介绍。
- 对三维模型的修改
当前市场上有不同的服务于三维影像建模的软件,其建模结果也根据软件的不同而存在差异。FDA建议用于将扫描图像转换为三维建模设计的软件需要对每个修改过程执行确认过程,并且确定设计人员手动进行设计改变的迭代过程。
-复杂的设计文件
人体的解剖结构是十分复杂的,包括那些曲线都在几何形状和数学记录方法上存在着极大的复杂性,这就为文件的每一次格式转换带来了挑战。为了被3D打印设备所读取,文件格式每被转化一次,就存在这一定的失真可能性。FDA在其增材制造医疗器械规范中重点介绍如何在文件转换过程中维护数据的一致性。
关于用于3D打印的数据资料需要被存档,并且需要参照通用的标准来进行识别。例如,一种方式是参考ISO/ASTM 52915在关于增材制造文件格式的标准规范-Standard specification for additive manufacturing file format (AMF)中关于文件格式的规范。而对于文件格式和文档控制系统来说,还需要增加材料和加工参数的表述,以使文件的完整性达到一致。
-网络安全和个人人体特征信息
对于患者的人体特征信息(PII-personally identifiable information)和保护健康信息(PHI-protected healthe information)需要依照HHS关于隐私规则重要方面的指导(HHS Guidance on Significant Aspects of the Privacy Rule).
从三维模型到3D打印设备
当完成建模后,经过处理的数据被发送到3D打印机进行制造过程。这个过程还包括很多环节,例如建构方向、支撑结构、切片、扫描策略等。
FDA的规范中指出在最终进行3D打印的医疗器械测试时,需要分析不同的步骤在3D打印过程中有哪些影响,若未对每个步骤有足够的理解,要找出是哪项制造缺失造成产品失败的根本原因就非常困难,因此建议在生产中应善用生产流程图,并在每个关键制造步骤中建立具建设性的提示与结论。
另外,即使以相同型号的3D打印设备,并以相同的制造参数、制造步骤和材料来生产相同的医疗器材或零件时,其生产产品的品质也有所不同。因此FDA也建议,制造商应清楚个输入参数和加工步骤的可变性,会如何影响终产品或零件的产生,这对于零件品质把关非常重要。若某步骤的结果无法通过后续检查和测试验证,则该过程必须执行严格的验证过程,并按既定的程序进行批准。
这些与加工有关的参数都需要被一一精确的记录下来,例如在铺粉的过程中,所选择的粉末层厚度也需要被记录下来,以分析其对产品性能、精度、质量和打印速度的影响。
而关于设备的硬件参数也需要被一一记录下来。在自动化和软件控制方面,还需要参考FDA的关于软件验证的一般原则(General Principle of Software Validation)的规范(资料下载,请加入3D科学谷3D产业链QQ群:529965687)。
![]() FDA的指导规范还对原材料表征、原材料生物相容性、医疗器械的质量监测方法(包括超声、CT、xx射线、共聚焦显微镜、高光谱成像等)、后处理(热等静压等)进行了详细的规范要求。
FDA的指导规范还对原材料表征、原材料生物相容性、医疗器械的质量监测方法(包括超声、CT、xx射线、共聚焦显微镜、高光谱成像等)、后处理(热等静压等)进行了详细的规范要求。
规范中也建议,通过3D打印制造的医疗器材在提交上市前审查时,应准备符合FDA要求的设备描述、机械测试、尺寸测量、材料表征、去除制造材料的残留物、杀菌和生物相容性等的文件。
美国FDA为了能够理解3D打印的技术本质,更以最先进的3D打印装置进行研究,以推动建立3D打印产品品质和安全的管理框架,推动法规科学的发展,如CDER药物评估及研究中心)关心3D打印如何影响药物非活性成分和其他药物成分,及制造过程的质量控制; CDRH(Centre for Devices and Radiological Health)则使用3D打印装置,研究医疗器材的制造方式变更是否对器材安全性和性能造成影响,以及每次产品的制造或仪器经迭代更新后,是否会改变产品的安全性和功效,例如义肢的适用性和舒适性是否因此改变。
FDA颁布这项规范,不仅为厂商指出更明确的监管架构,更有利于3D打印在医疗应用的发展,能够在确定的法律框架下进行开发,有助于医疗器材实现定制化、高品质开发。此外,美国FDA还计划审查与生物、细胞和组织产品相关的3D生物打印监管问题,以确定是否需要在于11月发布与再生医学产品相关之法规规范。这将为3D打印在伤口治疗,甚至替代器官等软体组织的发展方面给予更明确的方向。
参考资料:
- 科技新报
- Technical considerations for addtive manufactured medical devices- Guidance for industry and food and drug administration staff
资料下载,请加入3D科学谷3D产业链QQ群:529965687
查找往期文章,请登陆www.51shape.com,在首页搜索关键词
网站投稿请发送至editor@51shape.com